What is Cystic Fibrosis?
Cystic fibrosis (CF) is the most common fatal genetic disease affecting Canadian children and young adults. At present, there is no cure.
CF causes various effects on the body, but mainly affects the digestive system and lungs. The degree of CF severity differs from person to person, however, the persistence and ongoing infection in the lungs, with destruction of lungs and loss of lung function, will eventually lead to death in the majority of people with CF.
Typical complications caused by cystic fibrosis are:
- Difficulty digesting fats and proteins
- Malnutrition and vitamin deficiencies because of inability to absorb nutrients
- Progressive lung damage from chronic infections and aberrant inflammation
- CF related diabetes
- Sinus infections
It is estimated that one in every 3,600 children born in Canada has CF. More than 4,300 Canadian children, adolescents, and adults with cystic fibrosis attend specialized CF clinics.
CAUSES OF CYSTIC FIBROSIS
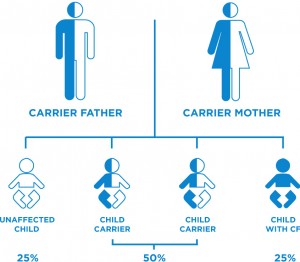
CF is a genetic disease that occurs when a child inherits two defective copies of the gene responsible for cystic fibrosis, one from each parent. Approximately, one in 25 Canadians carry one defective copy of the CF gene. Carriers do not have CF, nor do they exhibit any of the related symptoms.
When two CF carriers have a child, there is a 25 percent chance that the child will be born with CF. There is also a 50 percent chance that the child will be a carrier, and a 25 percent chance that the child will not be a carrier, nor have CF.
SYMPTOMS OF CYSTIC FIBROSIS
CF is a multi-system disorder that produces a variety of symptoms including:
- Persistent cough with productive thick mucous
- Wheezing and shortness of breath
- Frequent chest infections, which may include pneumonia
- Bowel disturbances, such as intestinal obstruction or frequent, oily stools
- Weight loss or failure to gain weight despite increased appetite
- Salty tasting sweat
- Infertility (men) and decreased fertility (women)
DIAGNOSING CYSTIC FIBROSIS
If a doctor suspects a patient has CF, a ‘sweat test’ may be administered. This test measures the amount of salt content present in the sweat. If the test comes back positive, it means the sweat collected contains more salt than usual and supports a diagnosis of CF. Genetic testing, prenatal and newborn screening for CF are other methods of determining the presence of CF.
